class: center, middle # BedTools Tools for interacting with genomic ranges --- #First, Tabular File Formats * [BED](https://genome.ucsc.edu/FAQ/FAQformat.html#format1) * [BED Detail](https://genome.ucsc.edu/FAQ/FAQformat.html#format1.7) - an extension of BED * [GFF](https://genome.ucsc.edu/FAQ/FAQformat.html#format3) - General Feature Format version 3 is best to follow [GFF3](http://www.sequenceontology.org/gff3.shtml) and the older [GFF2](http://www.sanger.ac.uk/resources/software/gff/spec.html) which is used in some places * [GTF](https://genome.ucsc.edu/FAQ/FAQformat.html#format4) - Gene Transfer Format --- #BED format ```code CHROM START END ``` Simple compact format to describe where a genomic feature is located. The first/leftmost value is '0' --- #GFF format More complex format ```code CHROM SOURCE TYPE START STOP STRAND SCORE PHASE GROUP ``` There are several variants on this format, mostly this takes place in how the GROUP column is encoded. GFF3 has a specific flavor of this while other formats have more relaxed guidelines. There can be genes, mRNA, CDS, exon, Untraslated region features. Group field can look like ```code ID=ENS0001;Name=Gene1 #or ID=ENST002;Parent=ENS001;Description="ensembl gene" ``` GTF format is a variant of GFF format where there are simple exons. It does not usually have the same Group fields --- #BEDTools "flexible tools with which to compare large sets of genomic features" It is intended for either manipulating or querying genomic features. The concept is that the genome is a linear coordinate system and you want to query intervals. It's goal is to be really fast at these computations. Here is the main website http://bedtools.readthedocs.org/en/latest/ --- #BEDTools 1. Intersect and Window queries 2. Extract fasta of features --- #BEDTools: Intersect and Window queries 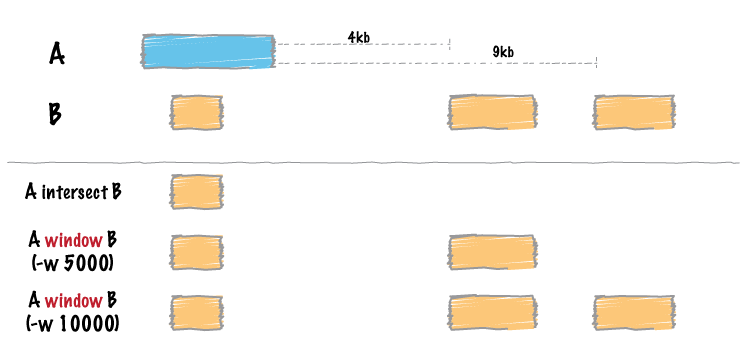 --- #BEDTools: Running intersect queries ```code $ bedtools intersect -a rice_random_exons.bed -b rice_random_exons.2.bed Chr3 12825367 12825459 Chr12 15129532 15129568 Chr4 31905111 31905385 Chr5 27970522 27971088 # use the -wa Write the original entry in A for each overlap. (default) # -wb Write the original entry in B for each overlap. # - Useful for knowing _what_ A overlaps. Restricted by -f and -r. $ bedtools intersect -a rice_random_exons.bed -b rice_random_exons.2.bed -wb Chr3 12825367 12825459 Chr3 12825367 12825459 Chr12 15129532 15129568 Chr12 15129532 15129568 Chr4 31905111 31905385 Chr4 31905111 31905385 Chr5 27970522 27971088 Chr5 27970522 27971088 # -r Require that the fraction overlap be reciprocal for A and B. # - In other words, if -f is 0.90 and -r is used, this requires # that B overlap 90% of A and A _also_ overlaps 90% of B. ``` --- #BEDTools: Running intersect/window queries ```shell # other options -wo # Write the original A and B entries plus the number of base # pairs of overlap between the two features. # - Overlapping features restricted by -f and -r. # However, A features w/o overlap are also reported # with a NULL B feature and overlap = 0. ``` --- #BEDTools: extract features from FASTA Common to have a set of locations defined in a file, this could be locations of CDS, exons, genes, SNPs, etc Could also be locations of protein domains on a protein sequence Want to extract these pieces as sequences in FASTA or other format --- #BEDTools: getfasta ```text $ bedtools getfasta Tool: bedtools getfasta (aka fastaFromBed) Version: v2.24.0 Summary: Extract DNA sequences into a fasta file based on feature coordinates. Usage: bedtools getfasta [OPTIONS] -fi <fasta> -bed <bed/gff/vcf> -fo <fasta> Options: -fi Input FASTA file -bed BED/GFF/VCF file of ranges to extract from -fi -fo Output file (can be FASTA or TAB-delimited) -name Use the name field for the FASTA header -split given BED12 fmt., extract and concatenate the sequences from the BED "blocks" (e.g., exons) -tab Write output in TAB delimited format. - Default is FASTA format. -s Force strandedness. If the feature occupies the antisense, strand, the sequence will be reverse complemented. - By default, strand information is ignored. -fullHeader Use full fasta header. - By default, only the word before the first space or tab is used ``` --- #BEDTools: Extract features with getfasta Two files * one is the fasta chromosome/genome file * one is the feature locations in BED/GFF/VCF format ```shell $ bedtools getfasta -fi Oryza_sativa.IRGSP-1.0.27.dna.chromosome.6.fa \ -bed Oryza_sativa.IRGSP-1.0.27.chromosome.6.gff3.gz -s \ -fo allfeats_chr6.fa # let's only get the CDS features $ zgrep CDS Oryza_sativa.IRGSP-1.0.27.chromosome.6.gff3.gz | \ bedtools getfasta -fi Oryza_sativa.IRGSP-1.0.27.dna.chromosome.6.fa \ -s -fo CDSfeats_chr6.fa -bed - ``` --- #BEDTools: extract SNPs and the flanking 50 bp around them Use the [slop tool](https://bedtools.readthedocs.org/en/latest/content/tools/slop.html) to expand window of the SNP. "bedtools slop will increase the size of each feature in a feature file by a user-defined number of bases. While something like this could be done with an ```code awk '{OFS="\t" print $1,$2-<slop>,$3+<slop>}' ``` bedtools slop will restrict the resizing to the size of the chromosome (i.e. no start < 0 and no end > chromosome size)." -- from the text slop needs the sizes of the chromosomes in order to not exceed the length. --- #BEDTools: extract the features ```shell # make a new bed file with larger flanking region $ bedtools slop -i rice_chr6_3kSNPs_filt.bed.gz -g chr6.genome.bed -b 50 > rice_chr6_3kSNPs_filt.flank.bed # then extract $ bedtools getfasta -fi Oryza_sativa.IRGSP-1.0.27.dna.chromosome.6.fa \ -bed rice_chr6_3kSNPs_filt.flank.bed -s -fo chr6_SNPs_flanking.fasta # remember you can actually do this all at once with pipes $ bedtools slop -i rice_chr6_3kSNPs_filt.bed.gz -g chr6.genome.bed -b 50 | \ bedtools getfasta -fi Oryza_sativa.IRGSP-1.0.27.dna.chromosome.6.fa -s \ -fo chr6_SNPs_flanking.fasta -bed - ``` --- #Tabix Another tool for interacting with ranged data. This is more suitable for random access to querying locations. __Data Files need to be Sorted__ Here's some simple code to sort it for you ```shell $ (grep ^"#" in.gff; grep -v ^"#" in.gff | sort -k1,1 -k4,4n) | bgzip > sorted.gff.gz # sort the data $ tabix -p gff sorted.gff.gz # build the index # now query for features in a range $ tabix sorted.gff.gz chr1:20000-40000 # here is example with data $ tabix rice_chr6_3kSNPs_filt.bed.gz # already sorted $ tabix rice_chr6_3kSNPs_filt.bed.gz 6:31217380-31236551 ``` --- #BEDTools - workshopping ```shell $ ssh pigeon.bioinfo.ucr.edu $ qsub -I $ module load bedtools # now try these examples out # all the data are in /bigdata/gen220/shared/GEN220_2015/data on cluster # or you can checkout the repository to your home or bigdata folder $ git clone https://github.com/hyphaltip/GEN220_2015.git $ cd GEN220_2015/data ```